
News|Articles|May 11, 2023
Genentech seeks indication for faricimab-svoa from the FDA for retinal vein inclusion (RVO)
Author(s)Kassi Jackson
If approved, RVO would be the third indication for faricimab-svoa in addition to wet age-related macular degeneration (AMD) and diabetic macular edema (DME).
Advertisement
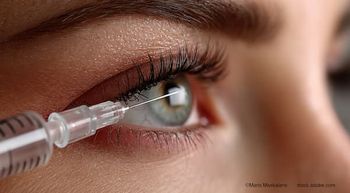
The US Food and Drug Administration (FDA) has accepted Genentech's supplemental Biologics License Application (sBLA) for faricimab-svoa (Vabysmo) for the treatment of macular edema following retinal vein occlusion (RVO).1
If approved by the FDA, faricimab-svoa would be approved for three treatments: wet
“This acceptance brings us one step closer to delivering Vabysmo as a treatment for retinal vein occlusion, a disease that affects more than one million people in the United States and can cause severe and sudden vision loss,” said Levi Garraway, MD, PhD, chief medical officer and head of Global Product Development. “If approved, this would be the third indication for Vabysmo, the first bispecific antibody available for the treatment of retinal conditions that can cause blindness.”
Affecting more than 1 million people in the US—mainly those aged 50 or older, RVO is the second most common cause of vision loss due to retinal vascular diseases. This sudden, painless vision loss in the affected eye is caused by vein blockage that restricts normal blood flow in the affected
The sBLA acceptance is based on results from the phase 3 BALATON and COMINO studies, which demonstrated that treatment with faricimab-svoa provides early and sustained improvement in vision after meeting the primary endpoint of non-inferior visual acuity gains at 24 weeks compared to aflibercept. Faricimab-svoa’s safety profile was consistent with previous trials and the data from the BALATON and COMINO studies will be submitted to other health authorities around the world, including the European Medicines Agency, for approval for the treatment of macular edema following RVO. The studies are ongoing, and data from weeks 24 to 72 will assess the potential of faricimab-svoa to extend dosing intervals up to every four months.
Faricimab-svoa safety and efficacy
Faricimab-svoa (Vabysmo) is the first bispecific antibody approved for the eye and was approved in the United States in January 2022 for the treatment of wet, or neovascular, age-related macular degeneration (AMD) and diabetic macular edema (DME). Wet AMD, DME, and RVO together affect around 3 million people in the US and are among the leading causes of vision loss.
Four global studies with more than 3,000 participants support the efficacy and safety profile of faricimab-svoa in wet AMD and DME; and it is the only injectable eye medicine approved for wet AMD and DME by the FDA with the option for treatments from 1 to 4 months apart in the first year following 4 initial monthly loading doses, based on evaluation of the patient’s anatomy and vision outcomes. It targets and inhibits 2 disease pathways linked to a number of vision-threatening retinal conditions by neutralizing angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).
There are 2 main types of RVO: branch retinal vein occlusion (BRVO), which affects an estimated 887,000 people in the U.S. and occurs when one of the four smaller “branches” of the main central retinal vein becomes blocked; and central retinal vein occlusion (CRVO), which is less common, affecting an estimated 265,000 people in the U.S., and occurs when the eye’s central retinal vein becomes blocked.
About the BALATON and COMINO studies
BALATON and COMINO are both randomized, multicenter, double-masked, global phase 3 studies evaluating the efficacy and safety of faricimab-svoa compared to aflibercept. For the first 20 weeks, patients are randomized 1:1 to receive 6 monthly injections of either faricimab-svoa (6.0 mg) or aflibercept (2.0 mg). From weeks 24 to 72, all patients receive faricimab-svoa (6.0 mg) up to every 4 months—according to a personalized treatment interval dosing regimen—using a treat-and-extend approach.
The BALATON study is being conducted in 553 patients with branch retinal vein occlusion. The COMINO study is being conducted in 729 patients with central retinal or hemiretinal vein occlusion.
The primary endpoint of each study is the change in best-corrected visual acuity (BCVA) from baseline at 24 weeks. Secondary endpoints include change in central subfield thickness (CST) from baseline over time up to 24 weeks.
Both studies met their primary endpoint, with faricimab-svoa showing non-inferior visual acuity gains compared to aflibercept. The average vision gains from baseline were comparable between the two treatments in both studies. In BALATON, vision gains were +16.9 eye chart letters in the faricimab-svoa arm and +17.5 letters in the aflibercept arm at 24 weeks. In COMINO, vision gains were +16.9 letters in the faricimab-svoa arm and +17.3 letters in the aflibercept arm at 24 weeks.
A secondary endpoint showed that faricimab-svoa achieved rapid and robust drying of retinal fluid, as measured by reduction in CST from baseline. In both studies, reductions in CST were comparable across treatment arms. In BALATON, CST reductions from baseline at 24 weeks were 311.4 μm in the faricimab-svoa arm and 304.4 μm in the aflibercept arm. In COMINO, CST reductions from baseline at 24 weeks were 461.6 μm in the faricimab-svoa arm and 448.8 μm in the aflibercept arm.
Additionally, both studies showed that more faricimab-svoa patients had an absence of blood vessel leakage in the retina compared to aflibercept patients as seen in a pre-specified exploratory endpoint. Blood vessel leakage in the macula may lead to more retinal fluid, which can cause swelling and blurry vision.
In BALATON, one-third of patients (34%) treated with faricimab-svoa had an absence of macular leakage compared to one-fifth (21%) of aflibercept patients at 24 weeks. In COMINO, the rates were 44% for faricimab-svoa patients versus 30% for aflibercept patients at 24 weeks.
In both studies, faricimab-svoa’s safety profile was consistent with previous trials. The most common adverse reaction was conjunctival hemorrhage (3%). Safety results were consistent across study arms.
The studies are ongoing, and data from weeks 24 to 72 will assess the potential of Vabysmo to extend dosing intervals up to every four months.
Reference(s)
Genentech: Press Releases | Monday, May 8, 2023. www.gene.com. Accessed May 11, 2023. https://www.gene.com/media/press-releases/14991/2023-05-08/fda-accepts-application-for-genentechs-v
Advertisement
Related Content
Advertisement



Latest CME

















Advertisement
Advertisement
Trending on Modern Retina
1
The Retina TL;DR with Dr. Weng: Navigating the ocular risks of facial fillers with Jennifer Murdock, MD
2
Retina World Congress 2026: Can vibration reduce pain during intravitreal injections?
3
Retina World Congress 2026: Sunir J. Garg, MD, FACS, on preventing occupational injury in retina practice
4
Retina World Congress 2026: Diagnosing macular telangiectasia in a field of mimickers
5