
|Articles|March 15, 2023
- Modern Retina Spring 2023
- Volume 3
- Issue 1
Developments in gene therapy for inherited optic neuropathies
Industry leaders look at what’s in the therapeutic delivery pipeline for these disorders.
Advertisement
By Benson S. Chen, MBChB, MSc, FRACP; Joshua P. Harvey, MA, BM BCh,
Pg Cert, FRCOphth; and Patrick Yu-Wai-Man, PhD, BMedSci, MBBS,
FRCPath, FRCOphth
Inherited optic neuropathies (IONs) are disorders that result in degeneration of the retinal ganglion cells (RGCs) and optic atrophy,1 affecting approximately 1 in 10,000 individuals in the general population. They represent an important cause of visual impairment and reduced quality of life in children and young adults. There are currently no effective treatments for most IONs.
However, the rapid pace of innovation within the fields of gene therapy and editing has accelerated the therapeutic delivery pipeline for these inherited forms of blindness. This article will provide an overview of recent developments in gene therapy for IONs and highlight some of the challenges unique to these disorders.
The clinical spectrum of IONs
The 2 most common IONs encountered in clinical practice are autosomal dominant optic atrophy (DOA) and Leber hereditary optic neuropathy (LHON).1 Historically, the diagnosis of both conditions was based on clinical characteristics including the patient’s demographic features, the pattern of vision loss, and the mode of inheritance. It is now recognized that both conditions are genetically heterogeneous with multiple genetic variants, causing the same clinical presentation.
DOA has an estimated prevalence of 1 in 25,000 in the United States.2 The disease is highly penetrant, with estimates of approximately 70% (43%-100%) reported.3 Typically, patients develop a bilateral optic atrophy that begins in the first 2 decades of life, resulting in progressive visual acuity and field loss that deteriorates to legal blindness in later life. Clinical management is limited to low-vision aids/rehabilitation, supportive care, and genetic/reproductive counseling.
LHON has an estimated prevalence of 1 in 30,000 to 50,000 in Northern Europe.4 In contrast to DOA, most individuals who carry a genetic mutation associated with LHON remain asymptomatic. Men who carry a LHON mitochondrial DNA (mtDNA) mutation are at greater risk of developing vision loss compared with women (17.5% vs 5.4%, respectively).5 Patients develop severe bilateral sequential or simultaneous vision loss (peak age at onset is 15-35 years) characterized by a dense central or cecocentral scotoma and visual acuity worse than 3.60 (Log MAR, 1.3). Idebenone (Raxone) is the only treatment for LHON authorized by the European Medicines Agency (EMA), with several clinical trials and real-world observational studies demonstrating benefit in a subgroup of treated patients.1
Although most patients with an ION experience isolated visual loss, some can develop more severe neurological features.
Approximately 20% of patients with DOA have evidence of extraocular features, which typically include a sensorineural hearing loss, peripheral neuropathy, and ataxia.1 Similarly, individuals affected by LHON can develop extraocular manifestations such as movement disorders or a multiple sclerosis–like syndrome (Harding disease).
Some multisystemic disorders, such as Wolfram syndrome and many of the primary mtDNA disorders, can also manifest with a DOA- or LHON-like vision loss and optic atrophy and, therefore, could be considered part of the clinical spectrum of IONs.
Gene therapy for autosomal DOA
The most common causative gene in DOA is OPA1 (3q21), which is responsible for more than 60% of DOA cases.6 OPA1 is critical in regulating mitochondrial fusion, bioenergetics, and cell death (apoptosis). OPA1 is
expressed in all cells in the body, but it is enriched in neural tissues and highly metabolically active organs such as the heart and liver. Why variants in OPA1 result specifically in loss of RGCs and no other cell types remains a subject of debate but may be caused by the unique structural and bioenergetic demands of RGCs and their particular gene expression pattern.6
Despite the challenges of developing genetic therapies for a gene that has many pathogenic variants, DOA remains an exciting target for gene therapies because it is autosomal (autosomal DNA is easier to edit than mtDNA); normally only affects 1 organ, which has an easy drug delivery route (intravitreal injection); and has a relatively gradual disease course, meaning there is—hypothetically—a large therapeutic window.
There are 2 broad strategies for gene therapy for DOA. The first is gene editing, and the second is modification of genetic expression by altering gene transcription. Gene editing is theoretically the most direct treatment of DOA, involving correction of the disease-causing OPA1 variant.
However, OPA1 is a relatively large gene (more than 90 kb), so a complete replacement gene cannot be packaged into an adeno-associated virus (AAV) vector. Gene editing strategies such as CRISPR-Cas9 have been used to correct OPA1 variants in vitro; however, the components of the system are again too large for an AAV vector, and concerns remain regarding low editing efficiency, off-target effects, and a potential risk of cell toxicity secondary to supraphysiological expression of OPA1.
An alternative approach that addresses these concerns involves the use of transcriptional modifiers such as antisense oligonucleotides (ASOs). ASOs are short portions of single-stranded nucleotides that can bind and modulate expression of mRNA or pre-mRNA. Stoke Therapeutics, Inc in Bedford, Massachusetts, is investigating an ASO that has been designed to upregulate functional OPA1 gene expression by downregulating nonfunctional/mutant transcript.
Another company, PYC Therapeutics in Perth, Australia, is trialing a related method for modulating gene expression called a peptide-conjugated phosphorodiamidate morpholino oligomer to restore levels of OPA1 expression. Transcriptional modification remains the most active area of translational research, and a number of these technologies are entering early-stage clinical trials. However, it remains to be seen if the promising preclinical data of increased OPA1 expression results in a clinically meaningful attenuation of RGC loss and visual benefit.
Gene therapy for LHON
Three primary point mutations in mtDNA (m.3460G>A in MT-ND1, m.11778G>A in MT-ND4, and m.14484T>C in MT-ND6) are responsible for approximately 90% of LHON cases globally.2
These mutations all involve genes encoding subunits of complex I—the first enzyme of the mitochondrial respiratory chain. In LHON, defective mitochondrial oxidative phosphorylation precipitates a bioenergetic crisis; oxidative damage to DNA, proteins, and lipids secondary to elevated levels of toxic reactive oxygen species; and release of signaling factors that trigger RGC apoptosis. Like DOA, most cases of LHON appear to cause selective degeneration of RGCs, especially the relatively smaller fibers that make up the papillomacular bundle.1
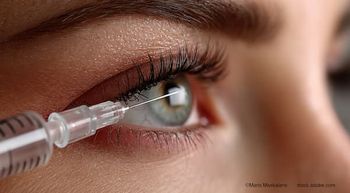
Because of the relatively impervious nature of the double mitochondrial membrane, conventional AAV vectors are prevented from entering the mitochondria or transferring exogenous genetic material into the mitochondrial matrix. Instead, gene therapy in LHON has utilized the technique of allotopic expression.
Several clinical trials have been conducted for the m.11778G>A mutation in MT-ND4, the most prevalent mutation causing LHON, accounting for 60% to 90% of cases, depending on the population surveyed.
Three separate groups (Bascom Palmer Eye Institute, Miami, Florida; Huazhong University of Science and Technology and Neurophth Therapeutics, Wuhan, China; and GenSight Biologics, Paris, France) have independently conducted gene therapy clinical trials. Although differences in treatment efficacy have been reported, possibly related to variations in study and vector design, gene therapy was found to be well tolerated across all studies, with transient ocular inflammation the main adverse effect identified.
GenSight Biologics has completed and published the results of its phase 3 trials: RESCUE (NCT02652767), REVERSE (NCT02652780), and REFLECT (NCT03293524).7,8,9 Eyes treated with lenadogene nolparvovec (Lumevoq) within 12-months onset of vision loss demonstrated a progressive and sustained improvement in best-corrected visual acuity from 12 to 51.5 months after onset of vision loss.
Compared with a natural history cohort, there was a statistically and clinically relevant difference in best-corrected visual acuity (improvement in 0.33 logMAR) in favor of treated eyes at 48 months after onset of vision loss.10 GenSight Biologics submitted a marketing authorization application for lenadogene nolparvovec to the EMA in September 2020, and an opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use is currently awaited.
Other gene therapy strategies under preclinical investigation are mtDNA heteroplasmy shifting and mitochondrial base editing. Mutant mtDNA molecules often exist in conjunction with the wild-type mtDNA species, a situation known as heteroplasmy. Mitochondrially targeted zinc finger nucleases and transcription activator–like effector nucleases have been used successfully in vitro and in animal models to induce heteroplasmic shift by favoring the replication of wild-type mtDNA molecules.11 However, this strategy has limited applicability for LHON because most carriers are homoplasmic mutant.
Another genomic approach being considered is mitochondrial base editing using non–CRISPR-based mtDNA editing tools, including DddA-derived cytosine base editor.12 The platform is still in its infancy, and it is currently limited to C•G-to-T•A editing, making it a viable strategy for the m.14484T>C mutation in MT-ND6.
Challenges to delivering gene therapy for IONs
Knowing whom to treat is critically important because treatments are likely to be expensive, requiring sometimes difficult decisions to be made by medical experts and policy makers.
A mutation-specific gene therapy treatment is unlikely to be available for all patients. Mutation-independent gene therapies that aim to improve mitochondrial respiration, reduce mitochondrial stress, inhibit or delay RGC apoptosis, and promote RGC survival are attractive because they can potentially be utilized in all patients with an ION in combination with other neuroprotective therapies, if available.
Specific genes under preclinical investigation that have been shown to improve defective mitochondrial function and/or increase RGC survival include SOD2, NRF2, and a novel transgene coding brain-derived neurotrophic factor and tropomyosin-related receptor B.13
Knowing when to treat is also important. For example, ASOs have a relatively short half-life. Timing a treatment or its frequency is likely to be critical in maximizing the therapeutic effect. DOA is a highly penetrant disease with a relatively gradual disease course, meaning there is hypothetically a large therapeutic window for all patients to be treated. Although gene therapy utilizing allotopic expression appears to be effective for patients with LHON treated within 1 year of onset of vision loss, the effect of gene therapy for patients with more chronic disease needs to be evaluated further. A better understanding of the natural history of IONs as well as prognostic factors may help stratify LHON carriers at highest risk of vision loss for prophylactic treatment when such an option becomes available.
Given that IONs are relatively rare conditions and that a potential therapy may not be appropriate for all patients, it is important to consider how the provision of these treatments can be financially viable, particularly in countries with less well-resourced health care systems.
Conclusion
Genetic therapies hold promise for IONs. With the number of therapies on the horizon for IONs, access to rapid genetic testing becomes even more important to establish the molecular diagnosis, aid visual prognostication, and help better understand genotype-phenotype correlations. •
References
1 Newman NJ, Yu-Wai-Man P, Biousse V, Carelli V. Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: towards improved diagnosis and management. Lancet Neurol. 2023;22(2):172-188. doi:10.1016/S1474-4422(22)00174-0
2 Yu-Wai-Man P, Griffiths PG, Burke A, et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010;117(8):1538-1546.e1. doi:10.1016/j.ophtha.2009.12.038
3 Lenaers G, Hamel C, Delettre C, et al. Dominant optic atrophy. Orphanet J Rare Dis. 2012;7:46. doi:10.1186/1750-1172-7-46
4 Chen BS, Yu-Wai-Man P. From bench to bedside-delivering gene therapy for Leber hereditary optic neuropathy. Cold Spring Harb Perspect Med. 2022;12(6):a041282. doi:10.1101/cshperspect.a041282
5 Lopez Sanchez MIG, Kearns LS, Staffieri SE, et al. Establishing risk of vision loss in Leber hereditary optic neuropathy. Am J Hum Genet. 2021;108(11):2159-2170. doi:10.1016/j.ajhg.2021.09.015
6 Yu-Wai-Man P, Chinnery PF. Dominant optic atrophy: novel OPA1 mutations and revised prevalence estimates Ophthalmology. 2013;120(8):1712-1712.e1. doi:10.1016/j.ophtha.2013.04.022
7 Yu-Wai-Man P, Newman NJ, Carelli V, et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med. 2020;12(573):eaaz7423. doi:10.1126/scitranslmed.aaz7423
8 Newman NJ, Yu-Wai-Man P, Carelli V, et al. Efficacy and safety of intravitreal gene therapy for Leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. 2021;128(5):649-660. doi:10.1016/j.ophtha.2020.12.012
9 Newman NJ, Yu-Wai-Man P, Subramanian PS, et al. Randomized trial of bilateral gene therapy injection for m.11778G > A MT-ND4 Leber optic neuropathy. Brain. Published online November 9, 2022. doi:10.1093/brain/awac421
10 Newman NJ, Yu-Wai-Man P, Carelli V, et al. Intravitreal gene therapy vs natural history in patients with Leber hereditary optic neuropathy carrying the m.11778G>A ND4 mutation: systematic review and indirect comparison. Front Neurol. 2021;12:662838. doi:10.3389/fneur.2021.662838
11 Jackson CB, Turnbull DM, Minczuk M, Gammage PA. Therapeutic manipulation of mtDNA heteroplasmy: a shifting perspective. Trends Mol Med. 2020;26(7):698-709. doi:10.1016/j.molmed.2020.02.006
12 Mok BY, de Moraes MH, Zeng J, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583(7817):631-637. doi:10.1038/s41586-020-2477-4
13 Osborne A, Khatib TZ, Songra L, et al. Neuroprotection of retinal ganglion cells by a novel gene therapy construct that achieves sustained enhancement of brain-derived neurotrophic factor/tropomyosin-related kinase receptor-B signaling. Cell Death Dis. 2018;9(10):1007. Published 2018 Sep 26. doi:10.1038/s41419-018-1041-8
Articles in this issue
about 3 years ago
Suprachoroidal TA for macular edema caused by noninfectious uveitisabout 3 years ago
Early detection and treatment of DR and DME are essentialabout 3 years ago
Vitrectomy: Is higher speed always better?about 3 years ago
IOL designed for AMD offers hope for patientsAdvertisement
Related Content
Advertisement



Latest CME

















Advertisement
Advertisement
Trending on Modern Retina
1
Retina World Congress 2026: Identifying real-world durability gaps in wet AMD treatment
2
Retina World Congress 2026: The case for earlier surgical intervention in diabetic macular edema
3
Retina World Congress 2026: Diagnosing macular telangiectasia in a field of mimickers
4
Retina World Congress 2026: Rethinking macular hole surgery without gas tamponade
5