
|Articles|February 12, 2018
FDA approval of retinal dystrophy drug launches era of ocular gene therapy
Author(s)Cheryl Guttman Krader, BS, Pharm
Voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics) was approved in December 2017 for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Clinical trial results and patient selection issues for this gene therapy are discussed.
Advertisement
Reviewed by Alex V. Levin, MD, and Paulo Falabella, MD
In December 2017, the FDA approved voretigene neparvovec-rzyl (“voretigene,” Luxturna, Spark Therapeutics), an adeno-associated virus serotype 2 vector-based gene therapy, for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy (RD).
With the regulatory agency’s decision, Luxturna became the first pharmacologic treatment for this inherited, progressive disease that often leads to nearly complete blindness and the first gene therapy in the United States indicated for treatment of a genetic disease. The treatment is expected to be available in March 2018.
Its approval was granted based on clinical trial results showing that the gene therapy had an acceptable safety profile and resulted in rapid improvements in functional vision and visual function that were sustained with follow-up to 2 years. Data reported at the 2017 American Academy of Ophthalmology meeting show the treatment benefit persists to at least 3 years.
“The approval heralds a new horizon for treatment of inherited RDs,” said Alex V. Levin, MD, MHSc, chief, pediatric ophthalmology and ocular genetics, Wills Eye Hospital, Philadelphia. “It behooves affected patients to get accurate molecular diagnosis of their underlying mutations so that they can bring themselves closer to intervention.”
“FDA approval of Luxturna is a landmark event that represents the culmination of nearly 20 years of hard work and a breakthrough for patients affected with biallelic RPE65 mutation-associated RD, their families, and the scientific community,” added Paulo Falabella, MD, medical affairs ophthalmic lead, Spark Therapeutics, Philadelphia. “Needless to say, Spark Therapeutics is very pleased and excited to bring it to market.”
Not a magic bullet
Dr. Levin pointed out that gene therapy with voretigene is not a magic bullet, even for patients who are appropriate candidates.
“We still have a lot to learn, and gene therapy needs to be more affordable and accessible,” Dr. Levin explained. “But the availability of this approved therapy that can deliver a replacement or repair mechanism for this one abnormal gene in the eye, halt progression of a retinal degeneration in some patients, and perhaps even restore sight brings us through a doorway to an exciting future.”
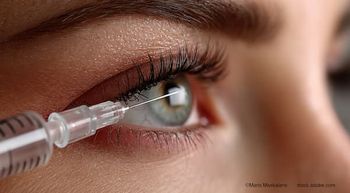
Voretigene is approved for use in patients aged ≥1 year who have viable retinal cells as determined by the treating physician. It is administered by subretinal injection (1.5 x 1011 vector genomes/0.3 mL) following vitrectomy.
Fellow eye procedures may be performed within a short timeframe, but with a minimum of six days between eyes. It is recommended that patients receive a systemic oral corticosteroid beginning three days prior to treatment to each eye. The recommended daily dose is prednisone equivalent 1 mg/kg (maximum 40 mg) for seven days followed by a taper during the next 10 days.
The treatment delivers a normal copy of the gene encoding the RPE65 protein to RPE cells. It thereby has the potential to restore the visual cycle and vision in individuals with RPE65 gene mutations, who have reduced or absent levels of RPE65.
Clinical trial results
The phase III clinical trial enrolled patients aged ≥4 years of age with confirmed biallelic mutations in the RPE65 gene, visual acuity <20/60 and/or visual field <20° in any meridian, and ability to perform a standardized multi-luminance mobility test (MLMT), within the evaluated luminance range, but unable to pass at 1 lux.
It randomized 31 patients 2:1 to voretigene or observation. Observation patients became eligible for crossover to gene therapy after 1 year, and in total 29 of the 31 entered patients received intervention and were evaluated at 3 years post-entry.
Change in functional vision–as measured by performance in the MLMT at 1-year post-injection–was analyzed as the primary endpoint. Change in visual function measured by white-light, full-field sensitivity was the secondary efficacy endpoint. Changes in visual field and best-corrected visual acuity (BCV) also were evaluated.
“Spark developed the MLMT in response to the need for a relevant, reliable, and clinically meaningful measure of functional vision in low-vision subjects with nyctalopia, but this novel endpoint had to be validated first in a separate study,” said Dr. Falabella.
“One of the challenges in developing voretigene was to establish an endpoint that could show improvement in functional vision.
“Visual acuity and visual field, which are traditional endpoints in ophthalmology trials, are visual function tests measuring how the eye performs,” he added. “A functional vision test measures how patients perform with the level of vision they have.”
Improvements in MLMT scores
The original intervention arm and the crossover patients who received delayed intervention showed statistically significant improvements in MLMT score, white-light full-field sensitivity, and visual field at the first outcome assessment on day 30 post-injection. The treatment benefits were sustained throughout follow-up.
At their last test, 69% of all subjects were able to pass the MLMT at the lowest light level. Light sensitivity improved by about 100-fold in the original intervention group and almost 500-fold in the delayed intervention group. Change in BCVA was not significantly different between the intervention and control groups.
“Improvements were reported 30 days after intervention,” said Dr. Falabella. Voretigene has the potential to provide lifelong efficacy because RPE cells are post-mitotic and unlikely to proliferate. Results so far support the expectation for sustained benefit, but, of course, only time will tell.”
In the Phase III trial, one ocular serious adverse event was reported in one eye, in which there was foveal thinning and a sustained reduction in BCVA. The most common ocular adverse events were transient-elevated intraocular pressure, cataracts, retinal tears, and retinal deposits. No deleterious immune responses were reported.
“Because the delivery procedure involves fluid-air exchange, patients also experience a decrease in vision for a few days,” Dr. Falabella said. “As the rods begin to capture more light, there were reports of early photophobia, but that was transient. The recommended minimum six-day interval between fellow eye injections allows for recovery of vision between procedures and for assessment of any potential complications or adverse events.”
Patient selection
RPE65 mutation-associated RD is estimated to affect 1,000 to 3,000 people in the United States. It can be diagnosed as Leber congenital amaurosis type 2, early onset severe retinal dystrophy, severe early childhood-onset retinal dystrophy, and retinitis pigmentosa type 20 among others.
Diagnostic confirmation through genetic testing is mandatory for identifying patients who would be eligible for voretigene. Spark Therapeutics is offering access to the evaluation.
“RPE65 mutation-associated RD presents with a spectrum of symptoms, which makes it difficult for anyone to establish the diagnosis clinically,” said Dr. Falabella.
“Ophthalmologists who suspect a patient has RD-affecting rods should refer that individual for genetic testing,” Dr. Falabella added. “Night blindness, which may manifest as light-seeking behavior in very young patients, has been consistently linked to RPE65-mutation associated RD.”
Entry into the premarketing clinical trial was limited to children aged ≥3 years as a practical consideration so enrolled participants had the ability to perform the study assessments. However, the gene therapy was approved for treatment of children aged ≥1 year, recognizing that RPE65-mutation associated RD is a progressive degenerative disease and that efficacy of the gene therapy depends on the presence of viable cells.
“It is important to treat affected patients as early as possible, given the progressive nature of the disease, although age was not a clear predictor of response in the clinical trial and even the oldest patient in the study showed significant improvement after intervention,” Dr. Falabella said. “Our youngest participant was 4 years old, and so we will see if there is any trend after younger patients are treated.”
Dr. Falabella added that for all eligible patients, the potential treatment benefit has to be balanced against the potential risks of the delivery procedure. The 1-year age limit considered that retinal cells are still undergoing cell proliferation during the first year of life, which would potentially cause dilution or loss of voretigene.
Treatment centers designated
Spark Therapeutics has designated select treatment centers to help ensure the quality of Luxturna from storing and handling to administration. Each treatment center is staffed with healthcare professionals, including retinal specialists, nurses, and genetic counselors, who have experience caring for patients with inherited retinal diseases. (listing available at
“Our focus is on optimizing patient safety, but providing patient access is also important,” Dr. Falabella. “We took geographic distribution into consideration when selecting the treatment sites.”
Treating surgeons and pharmacists responsible for preparing the product will be required to undergo educational programs sponsored by Spark Therapeutics.
In its commitment to help patients gain access to gene therapy, Spark Therapeutics’ patient support program is providing assistance to eligible patients with travel logistics and navigating the complexities of the reimbursement. Cost for Luxturna was set at $425,000 per eye.
Disclosures:
Alex V. Levin, MD, MHSc
E: alevin@willseye.org
Dr. Levin is a consultant to Spark Therapeutics
Paulo Falabella, MD
E: paulo.falabella@sparktx.com
Dr. Falabella is an employee of Spark Therapeutics and shareholder.
Advertisement
Related Content
Advertisement



Latest CME

















Advertisement
Advertisement
Trending on Modern Retina
1
Retina World Congress 2026: Identifying real-world durability gaps in wet AMD treatment
2
Retina World Congress 2026: The case for earlier surgical intervention in diabetic macular edema
3
Retina World Congress 2026: Diagnosing macular telangiectasia in a field of mimickers
4
Retina World Congress 2026: Rethinking macular hole surgery without gas tamponade
5